Compute the ground-state energy of formaldehyde using Fire Opal
Integrate Fire Opal into a quantum chemistry workflow for ground-state energy estimation
Computing the ground-state energy of a molecular system is a central task in electronic structure theory. From a classical perspective, high-accuracy methods such as Coupled Cluster with Singles and Doubles (CCSD) or multi-reference approaches can be used to obtain reliable reference energies, but their computational cost grows rapidly with system size and basis quality.
Quantum computing offers a complementary route, where you can map the molecular Hamiltonian to a qubit operator and use quantum circuits to approximate the ground state within the relevant fermionic Hilbert space. In this application note, you will solve the ground-state problem for formaldehyde ($\text{CH}_{2}\text{O}$), a small but chemically nontrivial molecule using Sample-based quantum diagonalization (SQD) eigenstate solver executed with and without Fire Opal. This application note covers the following:
- An introduction to the $\text{CH}_{2}\text{O}$ electronic structure problem and active-space construction
- An introduction to the SQD workflow
- Execution of the eigenstate solver for $\text{CH}_{2}\text{O}$
- Evaluation of the solver performance in terms of energy error
1. Introduction
You will compute the correlated ground-state energy of formaldehyde ($\text{CH}_{2}\text{O}$). The problem is formulated in a standard second-quantized setting, reduced to a chemically meaningful active space, and then mapped to a qubit Hamiltonian suitable for near-term processors. The Sample-based Quantum Diagonalization (SQD) solver uses bitstrings sampled from this Hamiltonian to reconstruct an approximate ground state. By executing the same SQD workflow with and without Fire Opal, you will obtain a controlled comparison of accuracy and robustness for a nontrivial, yet still classically tractable, molecular benchmark.
1.1 Electronic structure of formaldehyde ($\text{CH}_{2}\text{O}$)
Let's start with a standard quantum-chemistry pipeline. Given the formaldehyde's molecular geometry in the 3-21G basis set, a Hartree–Fock (HF) calculation provides a set of molecular orbitals, their occupations, and a mean-field ground-state energy. The Jordan–Wigner transformation is used to map the fermionic wavefunction of formaldehyde onto a qubit wavefunction that can be prepared on a quantum circuit. The JW mapping takes a system with $M$ spatial orbitals and represents it in a Hilbert space of $2M$ qubits, each spatial orbital is split into two spin orbitals, one for spin-up $\alpha$ and one for spin-down $\beta$, each represented by a single qubit.
For formaldehyde in the 3-21G basis, the Hartree–Fock calculation gives 22 spatial orbitals, $22\alpha+22\beta=44$ spin orbitals. Neutral $\text{CH}_{2}\text{O}$ has 16 electrons, and with a singlet reference $\text{spin}=0$ this corresponds to $8\alpha$ and $8\beta$ electrons. In the correlated calculation, the two lowest-energy spatial orbitals are frozen, they remain doubly occupied and do not participate in the calculation. This removes $4$ core electrons, leaving $12$ active electrons ($6\alpha$ and $6\beta$) distributed among the $20$ active spatial orbitals, or $40$ spin orbitals, so the quantum circuit effectively uses $40$ qubits to encode the electronic structure. The resulting fermionic Hilbert space has dimension $\binom{20}{6}^2 \approx 1.5 \times 10^9$, which is far too large for exact diagonalization.
1.2 Sample-based Quantum Diagonalization (SQD) workflow
To approximate the ground state of the $\text{CH}_{2}\text{O}$ Hamiltonian on a quantum device, the Sample-based Quantum Diagonalization (SQD) eigenstate solver is used, which combines a parameterized quantum circuit that explores relevant regions of Hilbert space with a classical subspace solver that reconstructs an approximate ground state from the sampled configurations. By working in a data-driven subspace determined directly from measurements on the device, SQD avoids the need to manipulate the full $2^N$-dimensional state vector while still capturing the dominant components of the ground state. The iterative nature of the method naturally provides diagnostics, such as orbital occupancy patterns, that can be used to assess whether the reconstructed state remains physically meaningful.
2. Imports and initializations
The following section sets up the necessary imports, credentials, and helper functions.
%pip install qiskit_addon_sqd qctrl-visualizer pyscffrom qiskit import QuantumCircuit, qasm3
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_addon_sqd.configuration_recovery import recover_configurations
from qiskit_addon_sqd.fermion import bitstring_matrix_to_ci_strs, solve_fermion
from qiskit_addon_sqd.subsampling import postselect_by_hamming_right_and_left, subsample
from qiskit_addon_sqd.counts import counts_to_arrays
import ffsim
from pyscf import cc
from pyscf.geomopt.geometric_solver import optimize
from pyscf import ao2mo, gto, mcscf, scf
import numpy as np
import fireopal as fo
import qctrlvisualizer as qv
import matplotlib.ticker as mticker
import matplotlib.pyplot as plt2.1 Credentials and backend
This notebook is developed to run on an IBM backend. To run it, you need an IBM Quantum account. Set up Fire Opal with your IBM account information and choose a backend.
token = "YOUR_IBM_CLOUD_API_KEY"
instance = "YOUR_IBM_CRN"
credentials = fo.credentials.make_credentials_for_ibm_cloud(
token=token, instance=instance
)Q-CTRL authentication successful!
# Enter your desired IBM backend here.
service = QiskitRuntimeService()
backend = service.backend("your_desired_backend")3. Molecular geometry and Hartree–Fock reference
You start by defining the formaldehyde molecule in a minimal but chemically meaningful setup. The Mole object specifies the nuclear geometry, the electronic structure model, and basic quantum numbers. The atomic positions correspond to a standard equilibrium geometry for $\text{CH}_{2}\text{O}$, with the oxygen and carbon placed along the $x$ axis and the two hydrogens arranged asymmetrically around the carbon, capturing the planar structure of the molecule. The total charge is set to zero and the spin multiplicity corresponds to a singlet state spin = 0, so the calculation describes neutral formaldehyde in its electronic ground state. A restricted Hartree–Fock (RHF) calculation is performed via scf.RHF(mol).run(). This provides the mean-field reference energy and the molecular orbital coefficients that are used in subsequent steps to construct the active-space Hamiltonian and its qubit representation.
# Formaldehyde
mol = gto.Mole()
mol.build(
verbose=0,
atom=[
["O", (0.6123, 0.0000, 0.0000)],
["C", (-0.6123, 0.0000, 0.0000)],
["H", (-1.2000, 0.2426, -0.8998)],
["H", (-1.2000, -0.2424, 0.8998)],
],
basis="3-21G",
spin=0,
charge=0,
symmetry=False,
)
mf = scf.RHF(mol).run()<pyscf.gto.mole.Mole at 0x7cf6e9250e10>
The optimize routine takes the mean-field object mf as input and performs a geometry optimization using translation–rotation internal coordinates (TRIC). The resulting object contains the optimized nuclear positions used as the starting point for constructing the correlated Hamiltonian in the chosen active space.
mol_opt = optimize(mf, tol_grad=3e-4, verbose=0) # Use geomeTRIC/TRIC under the hoodgeometric-optimize called with the following command line:
/home/nemo/miniconda3/envs/q-chemistry/lib/python3.11/site-packages/ipykernel_launcher.py --f=/run/user/1000/jupyter/runtime/kernel-v3bb13e301defbc9bd1453787936876a3100b92123.json
[91m())))))))))))))))/[0m
[91m())))))))))))))))))))))))),[0m
[91m*)))))))))))))))))))))))))))))))))[0m
[94m#,[0m [91m()))))))))/[0m [91m.)))))))))),[0m
[94m#%%%%,[0m [91m())))))[0m [91m.))))))))*[0m
[94m*%%%%%%,[0m [91m))[0m [93m..[0m [91m,))))))).[0m
[94m*%%%%%%,[0m [93m***************/.[0m [91m.)))))))[0m
[94m#%%/[0m [94m(%%%%%%,[0m [93m/*********************.[0m [91m)))))))[0m
[94m.%%%%%%#[0m [94m*%%%%%%,[0m [93m*******/,[0m [93m**********,[0m [91m.))))))[0m
[94m.%%%%%%/[0m [94m*%%%%%%,[0m [93m**[0m [93m********[0m [91m.))))))[0m
[94m##[0m [94m.%%%%%%/[0m [94m(%%%%%%,[0m [93m,******[0m [91m/)))))[0m
[94m%%%%%%[0m [94m.%%%%%%#[0m [94m*%%%%%%,[0m [92m,/////.[0m [93m******[0m [91m))))))[0m
[94m#%[0m [94m%%[0m [94m.%%%%%%/[0m [94m*%%%%%%,[0m [92m////////,[0m [93m*****/[0m [91m,)))))[0m
[94m#%%[0m [94m%%%[0m [94m%%%#[0m [94m.%%%%%%/[0m [94m(%%%%%%,[0m [92m///////.[0m [93m/*****[0m [91m))))).[0m
[94m#%%%%.[0m [94m%%%%%#[0m [94m/%%%%%%*[0m [94m#%%%%%%[0m [92m/////)[0m [93m******[0m [91m))))),[0m
[94m#%%%%##%[0m [94m%%%#[0m [94m.%%%%%%/[0m [94m(%%%%%%,[0m [92m///////.[0m [93m/*****[0m [91m))))).[0m
[94m##[0m [94m%%%[0m [94m.%%%%%%/[0m [94m*%%%%%%,[0m [92m////////.[0m [93m*****/[0m [91m,)))))[0m
[94m#%%%%#[0m [94m/%%%%%%/[0m [94m(%%%%%%[0m [92m/)/)//[0m [93m******[0m [91m))))))[0m
[94m##[0m [94m.%%%%%%/[0m [94m(%%%%%%,[0m [93m*******[0m [91m))))))[0m
[94m.%%%%%%/[0m [94m*%%%%%%,[0m [93m**.[0m [93m/*******[0m [91m.))))))[0m
[94m*%%%%%%/[0m [94m(%%%%%%[0m [93m********/*..,*/*********[0m [91m*))))))[0m
[94m#%%/[0m [94m(%%%%%%,[0m [93m*********************/[0m [91m)))))))[0m
[94m*%%%%%%,[0m [93m,**************/[0m [91m,))))))/[0m
[94m(%%%%%%[0m [91m()[0m [91m))))))))[0m
[94m#%%%%,[0m [91m())))))[0m [91m,)))))))),[0m
[94m#,[0m [91m())))))))))[0m [91m,)))))))))).[0m
[91m()))))))))))))))))))))))))))))))/[0m
[91m())))))))))))))))))))))))).[0m
[91m())))))))))))))),[0m
-=# [1;94m geomeTRIC started. Version: 1.1 [0m #=-
Current date and time: 2025-11-27 22:44:01
#========================================================#
#| [92m Arguments passed to driver run_optimizer(): [0m |#
#========================================================#
customengine <pyscf.geomopt.geometric_solver.PySCFEngine object at 0x7cf65895f890>
input /tmp/tmp2abw76ya/09ad8e40-f9ba-43da-99ef-02144fb147e9
logIni /home/nemo/miniconda3/envs/q-chemistry/lib/python3.11/site-packages/pyscf/geomopt/log.ini
maxiter 100
tol_grad 0.0003
verbose 0
----------------------------------------------------------
Custom engine selected.
Bonds will be generated from interatomic distances less than 1.20 times sum of covalent radii
12 internal coordinates being used (instead of 12 Cartesians)
Internal coordinate system (atoms numbered from 1):
Distance 1-2
Distance 2-3
Distance 2-4
Angle 1-2-4
Angle 3-2-4
Out-of-Plane 2-1-3-4
Translation-X 1-4
Translation-Y 1-4
Translation-Z 1-4
Rotation-A 1-4
Rotation-B 1-4
Rotation-C 1-4
<class 'geometric.internal.Distance'> : 3
<class 'geometric.internal.Angle'> : 2
<class 'geometric.internal.OutOfPlane'> : 1
<class 'geometric.internal.TranslationX'> : 1
<class 'geometric.internal.TranslationY'> : 1
<class 'geometric.internal.TranslationZ'> : 1
<class 'geometric.internal.RotationA'> : 1
<class 'geometric.internal.RotationB'> : 1
<class 'geometric.internal.RotationC'> : 1
> ===== Optimization Info: ====
> Job type: Energy minimization
> Maximum number of optimization cycles: 100
> Initial / maximum trust radius (Angstrom): 0.100 / 0.300
> Convergence Criteria:
> Will converge when all 5 criteria are reached:
> |Delta-E| < 1.00e-06
> RMS-Grad < 3.00e-04
> Max-Grad < 4.50e-04
> RMS-Disp < 1.20e-03
> Max-Disp < 1.80e-03
> === End Optimization Info ===
Step 0 : Gradient = 2.059e-02/3.166e-02 (rms/max) Energy = -113.2208012969
Hessian Eigenvalues: 4.50000e-02 5.00000e-02 5.00000e-02 ... 3.34877e-01 3.34925e-01 9.33739e-01
Step 1 : Displace = [0m1.763e-02[0m/[0m2.236e-02[0m (rms/max) Trust = 1.000e-01 (=) Grad = [0m1.628e-03[0m/[0m1.923e-03[0m (rms/max) E (change) = -113.2218079630 ([0m-1.007e-03[0m) Quality = [0m0.942[0m
Hessian Eigenvalues: 4.50019e-02 5.00000e-02 5.00000e-02 ... 3.34867e-01 3.65172e-01 9.26718e-01
Step 2 : Displace = [0m4.070e-03[0m/[0m5.007e-03[0m (rms/max) Trust = 1.414e-01 ([92m+[0m) Grad = [0m1.187e-03[0m/[0m1.689e-03[0m (rms/max) E (change) = -113.2218095419 ([0m-1.579e-06[0m) Quality = [0m0.102[0m
Hessian Eigenvalues: 4.58079e-02 5.00000e-02 5.00000e-02 ... 3.32929e-01 5.47468e-01 9.12700e-01
Step 3 : Displace = [0m2.002e-03[0m/[0m2.530e-03[0m (rms/max) Trust = 2.035e-03 ([91m-[0m) Grad = [0m4.283e-04[0m/[0m5.995e-04[0m (rms/max) E (change) = -113.2218191912 ([0m-9.649e-06[0m) Quality = [0m0.865[0m
Hessian Eigenvalues: 4.56858e-02 5.00000e-02 5.00000e-02 ... 3.33057e-01 5.89147e-01 9.40664e-01
Step 4 : Displace = [92m1.133e-03[0m/[92m1.690e-03[0m (rms/max) Trust = 2.878e-03 ([92m+[0m) Grad = [0m3.888e-04[0m/[0m6.601e-04[0m (rms/max) E (change) = -113.2218190143 ([92m+1.769e-07[0m) Quality = [91m-0.148[0m
Hessian Eigenvalues: 4.99980e-02 5.00000e-02 5.00000e-02 ... 3.31688e-01 4.22814e-01 9.42406e-01
Step 5 : Displace = [92m5.493e-04[0m/[92m8.873e-04[0m (rms/max) Trust = 5.666e-04 ([91m-[0m) Grad = [92m1.596e-04[0m/[92m2.191e-04[0m (rms/max) E (change) = -113.2218199050 ([92m-8.907e-07[0m) Quality = [0m0.805[0m
Hessian Eigenvalues: 4.99980e-02 5.00000e-02 5.00000e-02 ... 3.31688e-01 4.22814e-01 9.42406e-01
Converged! =D
#==========================================================================#
#| If this code has benefited your research, please support us by citing: |#
#| |#
#| Wang, L.-P.; Song, C.C. (2016) "Geometry optimization made simple with |#
#| translation and rotation coordinates", J. Chem, Phys. 144, 214108. |#
#| http://dx.doi.org/10.1063/1.4952956 |#
#==========================================================================#
Time elapsed since start of run_optimizer: 15.797 seconds
With the optimized geometry you can recompute the restricted Hartree–Fock energy. To obtain a high-quality classical reference for the correlated ground-state energy, you can perform a CCSD calculation on the optimized Hartree–Fock reference, using a frozen-core approximation consistent with the active-space setup:
# Run the kernel to get the RHF energy
mf_opt = scf.RHF(mol_opt)
hf_e = float(mf_opt.kernel())
print(f"Restricted Hartree-Fock Energy: {hf_e}")Restricted Hartree-Fock Energy: -113.22181990503708
ccsd = cc.CCSD(mf_opt).set(frozen=2).run()
exact_energy = ccsd.e_tot
print("CCSD reference energy:", exact_energy)CCSD reference energy: -113.44730072803931
3.1 Active-space Hamiltonian and reference amplitudes
With the active-space definition, you can construct the corresponding many-body Hamiltonian and extract the integrals needed for the SQD solver.
open_shell = False
spin_sq = 0
n_frozen = 2
active_space = range(n_frozen, mol_opt.nao_nr())
num_orbitals = len(active_space)
n_electrons = int(sum(mf_opt.mo_occ[active_space]))
num_elec_a = (n_electrons + mol_opt.spin) // 2
num_elec_b = (n_electrons - mol_opt.spin) // 2
cas = mcscf.CASCI(mf_opt, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
t1 = ccsd.t1
t2 = ccsd.t2To generate a trial state for the SQD workflow, you can use a unitary cluster Jastrow (UCJ) ansatz constructed from the CCSD single and double excitation amplitudes. The ffsim library provides a convenient interface for this through the UCJOpSpinBalanced operator, which builds a spin-balanced UCJ circuit tailored to the active-space Hamiltonian.
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)4. Circuit preparation
With the active-space Hamiltonian and the UCJ operator defined, you can now build the quantum circuit that prepares the trial state used for sampling in the SQD workflow. The circuit acts on a register of $2\,\text{num\_orbitals}$ qubits, corresponding to the $\alpha$ and $\beta$ spin orbitals in the active space, and is initialized in the Hartree–Fock configuration before the UCJ transformation is applied.
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()pass_manager = generate_preset_pass_manager(optimization_level=3, backend=backend)
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")Pass: ContainsInstruction - 0.02122 (ms)Pass: UnitarySynthesis - 0.02456 (ms)Pass: HighLevelSynthesis - 101.91250 (ms)Pass: BasisTranslator - 0.17643 (ms)Pass: ElidePermutations - 0.01192 (ms)Pass: RemoveDiagonalGatesBeforeMeasure - 0.03171 (ms)Pass: RemoveIdentityEquivalent - 0.17881 (ms)Pass: InverseCancellation - 0.15092 (ms)Pass: ContractIdleWiresInControlFlow - 0.00715 (ms)Pass: CommutativeCancellation - 0.96846 (ms)Pass: ConsolidateBlocks - 8.52537 (ms)Pass: Split2QUnitaries - 0.01407 (ms)Pass: SetLayout - 0.00668 (ms)Pass: VF2Layout - 1.28508 (ms)Pass: BarrierBeforeFinalMeasurements - 0.51451 (ms)Pass: SabreLayout - 487.84590 (ms)Pass: CheckMap - 0.17428 (ms)Pass: VF2PostLayout - 1086.56836 (ms)Pass: ApplyLayout - 0.75722 (ms)Pass: FilterOpNodes - 0.62156 (ms)Pass: UnitarySynthesis - 0.01669 (ms)Pass: HighLevelSynthesis - 0.23365 (ms)Pass: BasisTranslator - 23.53239 (ms)Pass: Depth - 6.21009 (ms)Pass: Size - 0.01383 (ms)Pass: MinimumPoint - 0.01907 (ms)Pass: ConsolidateBlocks - 34.10459 (ms)Pass: UnitarySynthesis - 59.82471 (ms)Pass: RemoveIdentityEquivalent - 1.95932 (ms)Pass: Optimize1qGatesDecomposition - 14.50920 (ms)Pass: CommutativeCancellation - 16.81304 (ms)Pass: ContractIdleWiresInControlFlow - 0.01884 (ms)Pass: GatesInBasis - 2.93207 (ms)Pass: Depth - 3.53217 (ms)Pass: Size - 0.01526 (ms)Pass: MinimumPoint - 2.89083 (ms)Pass: ConsolidateBlocks - 41.71515 (ms)Pass: UnitarySynthesis - 23.73886 (ms)Pass: RemoveIdentityEquivalent - 0.95725 (ms)Pass: Optimize1qGatesDecomposition - 27.32229 (ms)Pass: CommutativeCancellation - 32.26256 (ms)Pass: ContractIdleWiresInControlFlow - 0.00978 (ms)Pass: GatesInBasis - 3.34454 (ms)Pass: Depth - 6.27613 (ms)Pass: Size - 0.01788 (ms)Pass: MinimumPoint - 13.30924 (ms)Pass: ConsolidateBlocks - 37.79292 (ms)Pass: UnitarySynthesis - 7.25460 (ms)Pass: RemoveIdentityEquivalent - 1.92499 (ms)Pass: Optimize1qGatesDecomposition - 9.87267 (ms)Pass: CommutativeCancellation - 15.27667 (ms)Pass: ContractIdleWiresInControlFlow - 0.01073 (ms)Pass: GatesInBasis - 2.11143 (ms)Pass: Depth - 2.46692 (ms)Pass: Size - 0.01550 (ms)Pass: MinimumPoint - 2.89059 (ms)Pass: ConsolidateBlocks - 22.40729 (ms)Pass: UnitarySynthesis - 7.41577 (ms)Pass: RemoveIdentityEquivalent - 0.94128 (ms)Pass: Optimize1qGatesDecomposition - 21.10672 (ms)Pass: CommutativeCancellation - 25.22111 (ms)Pass: ContractIdleWiresInControlFlow - 0.01431 (ms)Pass: GatesInBasis - 2.78139 (ms)Pass: Depth - 2.81262 (ms)Pass: Size - 0.01788 (ms)Pass: MinimumPoint - 0.01526 (ms)Pass: ConsolidateBlocks - 36.59749 (ms)Pass: UnitarySynthesis - 7.60293 (ms)Pass: RemoveIdentityEquivalent - 0.94342 (ms)Pass: Optimize1qGatesDecomposition - 7.11513 (ms)Pass: CommutativeCancellation - 29.15645 (ms)Pass: ContractIdleWiresInControlFlow - 0.01144 (ms)Pass: GatesInBasis - 2.70319 (ms)Pass: Depth - 9.57656 (ms)Pass: Size - 0.01335 (ms)Pass: MinimumPoint - 10.06651 (ms)Pass: ConsolidateBlocks - 50.69804 (ms)Pass: UnitarySynthesis - 17.57908 (ms)Pass: RemoveIdentityEquivalent - 1.05786 (ms)Pass: Optimize1qGatesDecomposition - 9.01985 (ms)Pass: CommutativeCancellation - 13.41367 (ms)Pass: ContractIdleWiresInControlFlow - 0.01240 (ms)Pass: GatesInBasis - 4.43721 (ms)Pass: Depth - 2.65527 (ms)Pass: Size - 0.01693 (ms)Pass: MinimumPoint - 0.01407 (ms)Pass: ConsolidateBlocks - 59.45563 (ms)Pass: UnitarySynthesis - 16.52598 (ms)Pass: RemoveIdentityEquivalent - 1.65534 (ms)Pass: Optimize1qGatesDecomposition - 22.98403 (ms)Pass: CommutativeCancellation - 33.03242 (ms)Pass: ContractIdleWiresInControlFlow - 0.01407 (ms)Pass: GatesInBasis - 1.99866 (ms)Pass: Depth - 7.59840 (ms)Pass: Size - 0.02432 (ms)Pass: MinimumPoint - 0.02217 (ms)Pass: ConsolidateBlocks - 34.21283 (ms)Pass: UnitarySynthesis - 18.53347 (ms)Pass: RemoveIdentityEquivalent - 0.92459 (ms)Pass: Optimize1qGatesDecomposition - 24.76335 (ms)Pass: CommutativeCancellation - 30.29871 (ms)Pass: ContractIdleWiresInControlFlow - 0.01121 (ms)Pass: GatesInBasis - 5.64694 (ms)Pass: Depth - 10.28943 (ms)Pass: Size - 0.01884 (ms)Pass: MinimumPoint - 0.01216 (ms)Pass: ConsolidateBlocks - 62.38580 (ms)Pass: UnitarySynthesis - 13.87620 (ms)Pass: RemoveIdentityEquivalent - 4.18258 (ms)Pass: Optimize1qGatesDecomposition - 29.88338 (ms)Pass: CommutativeCancellation - 9.11307 (ms)Pass: ContractIdleWiresInControlFlow - 0.00882 (ms)Pass: GatesInBasis - 2.06900 (ms)Pass: Depth - 3.16930 (ms)Pass: Size - 0.03076 (ms)Pass: MinimumPoint - 0.01645 (ms)Pass: VF2PostLayout - 781.11506 (ms)Pass: ContainsInstruction - 0.02766 (ms)Pass: InstructionDurationCheck - 0.01121 (ms)Pass: Decompose - 3.00145 (ms)Pass: MergeOrbitalRotations - 1.75023 (ms)Pass: UnitarySynthesis - 0.01740 (ms)Pass: HighLevelSynthesis - 41.18633 (ms)Pass: BasisTranslator - 0.16212 (ms)Pass: ElidePermutations - 0.01574 (ms)Pass: RemoveDiagonalGatesBeforeMeasure - 0.03266 (ms)Pass: RemoveIdentityEquivalent - 0.10657 (ms)Pass: InverseCancellation - 0.12231 (ms)Pass: ContractIdleWiresInControlFlow - 0.00644 (ms)Pass: CommutativeCancellation - 0.60129 (ms)Pass: ConsolidateBlocks - 2.79880 (ms)Pass: Split2QUnitaries - 0.01788 (ms)Pass: SetLayout - 0.01049 (ms)Pass: VF2Layout - 1.20568 (ms)Pass: BarrierBeforeFinalMeasurements - 0.42725 (ms)
Gate counts (w/o pre-init passes): OrderedDict([('sx', 8254), ('rz', 6923), ('cz', 3042), ('x', 500), ('measure', 40), ('barrier', 1)])
Pass: SabreLayout - 510.85472 (ms)Pass: CheckMap - 0.19765 (ms)Pass: VF2PostLayout - 175.64106 (ms)Pass: ApplyLayout - 0.82803 (ms)Pass: FilterOpNodes - 0.36907 (ms)Pass: UnitarySynthesis - 0.01335 (ms)Pass: HighLevelSynthesis - 0.15497 (ms)Pass: BasisTranslator - 8.40044 (ms)Pass: Depth - 2.19154 (ms)Pass: Size - 0.01073 (ms)Pass: MinimumPoint - 0.02050 (ms)Pass: ConsolidateBlocks - 15.45191 (ms)Pass: UnitarySynthesis - 26.01624 (ms)Pass: RemoveIdentityEquivalent - 0.71621 (ms)Pass: Optimize1qGatesDecomposition - 6.10590 (ms)Pass: CommutativeCancellation - 5.45287 (ms)Pass: ContractIdleWiresInControlFlow - 0.00978 (ms)Pass: GatesInBasis - 1.36399 (ms)Pass: Depth - 1.27196 (ms)Pass: Size - 0.00930 (ms)Pass: MinimumPoint - 1.31655 (ms)Pass: ConsolidateBlocks - 9.19533 (ms)Pass: UnitarySynthesis - 3.64876 (ms)Pass: RemoveIdentityEquivalent - 0.43750 (ms)Pass: Optimize1qGatesDecomposition - 3.54743 (ms)Pass: CommutativeCancellation - 5.38278 (ms)Pass: ContractIdleWiresInControlFlow - 0.00906 (ms)Pass: GatesInBasis - 1.37377 (ms)Pass: Depth - 1.03211 (ms)Pass: Size - 0.00739 (ms)Pass: MinimumPoint - 1.46317 (ms)Pass: ConsolidateBlocks - 8.11553 (ms)Pass: UnitarySynthesis - 3.71265 (ms)Pass: RemoveIdentityEquivalent - 0.45276 (ms)Pass: Optimize1qGatesDecomposition - 3.38793 (ms)Pass: CommutativeCancellation - 3.64780 (ms)Pass: ContractIdleWiresInControlFlow - 0.01025 (ms)Pass: GatesInBasis - 1.20711 (ms)Pass: Depth - 1.38736 (ms)Pass: Size - 0.01264 (ms)Pass: MinimumPoint - 2.06351 (ms)Pass: ConsolidateBlocks - 8.32605 (ms)Pass: UnitarySynthesis - 3.51977 (ms)Pass: RemoveIdentityEquivalent - 0.43988 (ms)Pass: Optimize1qGatesDecomposition - 3.06153 (ms)Pass: CommutativeCancellation - 3.60107 (ms)Pass: ContractIdleWiresInControlFlow - 0.00644 (ms)Pass: GatesInBasis - 1.19758 (ms)Pass: Depth - 0.95987 (ms)Pass: Size - 0.00930 (ms)Pass: MinimumPoint - 0.01168 (ms)Pass: VF2PostLayout - 1602.52237 (ms)Pass: ContainsInstruction - 0.02789 (ms)Pass: InstructionDurationCheck - 0.03958 (ms)
Gate counts (w/ pre-init passes): OrderedDict([('sx', 4555), ('rz', 3791), ('cz', 1646), ('x', 142), ('measure', 40), ('barrier', 1)])
sampler = Sampler(mode=backend)
job = sampler.run([isa_circuit], shots=10_000)base_primitive._run:INFO:2025-11-27 22:47:29,267: Submitting job using options {'options': {}, 'version': 2, 'support_qiskit': True}
5. Running the algorithm
Run the $\text{CH}_{2}\text{O}$ UCJ trial circuit, with and without Fire Opal, to collect the measurement samples required for the SQD ground-state reconstruction.
# Run cell after IQX job completion
primitive_result = job.result()
pub_result = primitive_result[0]
counts_default = pub_result.data.meas.get_counts()fire_opal_job = fo.execute(
circuits=[qasm3.dumps(circuit.decompose(reps=5))],
shot_count=10_000,
credentials=credentials,
backend_name=backend.name,
)counts_fo = fire_opal_job.result()["results"][0]6. SQD analysis and performance evaluation
The measurement data are now used as input to the SQD workflow. The function calculate_energies implements the full configuration-recovery and eigenstate-reconstruction loop: it converts the raw counts into bitstring and probability arrays, iteratively refines the configuration set using average orbital occupancies, and solves a sequence of reduced eigenvalue problems in the active-space sector.
rng = np.random.default_rng(24)
iterations = 3
samples_schedule = [600, 800, 1000, 1200, 1400]
experiments = len(samples_schedule)
n_batches = 5
max_davidson_cycles = 300def calculate_energies(counts):
bitstring_matrix_full, probs_arr_full = counts_to_arrays(counts)
e_hist = np.zeros((experiments, iterations, n_batches))
s_hist = np.zeros((experiments, iterations, n_batches))
occupancy_hist = [[None for _ in range(iterations)] for _ in range(experiments)]
for ex, spb in enumerate(samples_schedule):
print(f"\n=== Experiment {ex} – samples_per_batch = {spb} ===")
avg_occupancy = None
for i in range(iterations):
print(f" Starting configuration recovery iteration {i}")
if avg_occupancy is None:
bs_mat_tmp = bitstring_matrix_full
probs_arr_tmp = probs_arr_full
else:
bs_mat_tmp, probs_arr_tmp = recover_configurations(
bitstring_matrix_full,
probs_arr_full,
avg_occupancy,
num_elec_a,
num_elec_b,
rand_seed=rng,
)
postselected_bs_mat, postselected_probs_arr = (
postselect_by_hamming_right_and_left(
bs_mat_tmp,
probs_arr_tmp,
hamming_right=num_elec_a,
hamming_left=num_elec_b,
)
)
batches = subsample(
postselected_bs_mat,
postselected_probs_arr,
samples_per_batch=spb,
num_batches=n_batches,
rand_seed=rng,
)
e_tmp = np.zeros(n_batches)
s_tmp = np.zeros(n_batches)
occs_tmp = []
coeffs = []
for j in range(n_batches):
strs_a, strs_b = bitstring_matrix_to_ci_strs(batches[j])
print(f" Batch {j} subspace dimension: {len(strs_a) * len(strs_b)}")
energy_sci, coeffs_sci, avg_occs, spin = solve_fermion(
batches[j],
hcore,
eri,
open_shell=open_shell,
spin_sq=spin_sq,
max_cycle=max_davidson_cycles,
)
energy_sci += nuclear_repulsion_energy
e_tmp[j] = energy_sci
print(f" Energy {j}: {energy_sci}")
s_tmp[j] = spin
occs_tmp.append(avg_occs)
coeffs.append(coeffs_sci)
avg_occupancy = tuple(np.mean(occs_tmp, axis=0))
e_hist[ex, i, :] = e_tmp
s_hist[ex, i, :] = s_tmp
occupancy_hist[ex][i] = avg_occupancy
min_e = [np.min(e_hist[ex, -1, :]) for ex in range(experiments)]
e_diff = [abs(e - exact_energy) for e in min_e]
x1 = samples_schedule
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4, 1e-5]
chem_accuracy = 0.001
last_occ = occupancy_hist[-1][-1]
y2 = last_occ[0] + last_occ[1]
x2 = range(len(y2))
return e_hist, min_e, e_diff, yt1, chem_accuracy, x1, y2, x2# Convert counts into bitstring and probability arrays
e_hist_fo, min_e_fo, e_diff_fo, yt1_fo, chem_accuracy_fo, x1_fo, y2_fo, x2_fo = (
calculate_energies(counts_fo)
)=== Experiment 0 – samples_per_batch = 600 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 26896
Energy 0: -113.2875885778956
Batch 1 subspace dimension: 26896
Energy 1: -113.2875885778956
Batch 2 subspace dimension: 26896
Energy 2: -113.2875885778956
Batch 3 subspace dimension: 26896
Energy 3: -113.2875885778956
Batch 4 subspace dimension: 26896
Energy 4: -113.2875885778956
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 944784
Energy 0: -113.38930801929412
Batch 1 subspace dimension: 887364
Energy 1: -113.38744860524812
Batch 2 subspace dimension: 960400
Energy 2: -113.35849912053587
Batch 3 subspace dimension: 944784
Energy 3: -113.39705880577881
Batch 4 subspace dimension: 917764
Energy 4: -113.39633464939948
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 938961
Energy 0: -113.37303988498914
Batch 1 subspace dimension: 919681
Energy 1: -113.39721546711228
Batch 2 subspace dimension: 933156
Energy 2: -113.37750594584291
Batch 3 subspace dimension: 898704
Energy 3: -113.40958540942141
Batch 4 subspace dimension: 917764
Energy 4: -113.39255176437467
=== Experiment 1 – samples_per_batch = 800 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 26896
Energy 0: -113.2875885778956
Batch 1 subspace dimension: 26896
Energy 1: -113.2875885778956
Batch 2 subspace dimension: 26896
Energy 2: -113.2875885778956
Batch 3 subspace dimension: 26896
Energy 3: -113.2875885778956
Batch 4 subspace dimension: 26896
Energy 4: -113.2875885778956
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 1597696
Energy 0: -113.39304930080205
Batch 1 subspace dimension: 1505529
Energy 1: -113.42053694645612
Batch 2 subspace dimension: 1547536
Energy 2: -113.41426853784367
Batch 3 subspace dimension: 1565001
Energy 3: -113.40597830168264
Batch 4 subspace dimension: 1580049
Energy 4: -113.41215277160218
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 1633284
Energy 0: -113.41457064842888
Batch 1 subspace dimension: 1560001
Energy 1: -113.39599631150583
Batch 2 subspace dimension: 1587600
Energy 2: -113.41324919431219
Batch 3 subspace dimension: 1525225
Energy 3: -113.41421022080439
Batch 4 subspace dimension: 1535121
Energy 4: -113.40608012090325
=== Experiment 2 – samples_per_batch = 1000 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 26896
Energy 0: -113.2875885778956
Batch 1 subspace dimension: 26896
Energy 1: -113.2875885778956
Batch 2 subspace dimension: 26896
Energy 2: -113.2875885778956
Batch 3 subspace dimension: 26896
Energy 3: -113.2875885778956
Batch 4 subspace dimension: 26896
Energy 4: -113.2875885778956
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 2223081
Energy 0: -113.41988799271988
Batch 1 subspace dimension: 2268036
Energy 1: -113.40607217363949
Batch 2 subspace dimension: 2368521
Energy 2: -113.40585572265165
Batch 3 subspace dimension: 2280100
Energy 3: -113.41236684677321
Batch 4 subspace dimension: 2328676
Energy 4: -113.42128937377414
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 2353156
Energy 0: -113.42244456256088
Batch 1 subspace dimension: 2365444
Energy 1: -113.41463436757597
Batch 2 subspace dimension: 2347024
Energy 2: -113.41897041893098
Batch 3 subspace dimension: 2295225
Energy 3: -113.40180814803554
Batch 4 subspace dimension: 2304324
Energy 4: -113.38958960167072
=== Experiment 3 – samples_per_batch = 1200 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 26896
Energy 0: -113.2875885778956
Batch 1 subspace dimension: 26896
Energy 1: -113.2875885778956
Batch 2 subspace dimension: 26896
Energy 2: -113.2875885778956
Batch 3 subspace dimension: 26896
Energy 3: -113.2875885778956
Batch 4 subspace dimension: 26896
Energy 4: -113.2875885778956
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 3193369
Energy 0: -113.42801100400098
Batch 1 subspace dimension: 3214849
Energy 1: -113.4269231759451
Batch 2 subspace dimension: 3236401
Energy 2: -113.42484717301488
Batch 3 subspace dimension: 3189796
Energy 3: -113.42242835841776
Batch 4 subspace dimension: 3243601
Energy 4: -113.42542021227425
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 3374569
Energy 0: -113.42764822117307
Batch 1 subspace dimension: 3211264
Energy 1: -113.41503294551734
Batch 2 subspace dimension: 3308761
Energy 2: -113.42622169591407
Batch 3 subspace dimension: 3139984
Energy 3: -113.42485515013414
Batch 4 subspace dimension: 3268864
Energy 4: -113.42365017975288
=== Experiment 4 – samples_per_batch = 1400 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 26896
Energy 0: -113.2875885778956
Batch 1 subspace dimension: 26896
Energy 1: -113.2875885778956
Batch 2 subspace dimension: 26896
Energy 2: -113.2875885778956
Batch 3 subspace dimension: 26896
Energy 3: -113.2875885778956
Batch 4 subspace dimension: 26896
Energy 4: -113.28758857789562
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 4268356
Energy 0: -113.4191921027971
Batch 1 subspace dimension: 4116841
Energy 1: -113.42314780175872
Batch 2 subspace dimension: 4153444
Energy 2: -113.42741127130701
Batch 3 subspace dimension: 4048144
Energy 3: -113.42628121795349
Batch 4 subspace dimension: 4120900
Energy 4: -113.42862408342738
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 4145296
Energy 0: -113.42754883432441
Batch 1 subspace dimension: 4133089
Energy 1: -113.4254154603791
Batch 2 subspace dimension: 4289041
Energy 2: -113.42883832619535
Batch 3 subspace dimension: 4129024
Energy 3: -113.42193330941731
Batch 4 subspace dimension: 4243600
Energy 4: -113.4281623463776
(
e_hist_default,
min_e_default,
e_diff_default,
yt1_default,
chem_accuracy_default,
x1_default,
y2_default,
x2_default,
) = calculate_energies(counts_default)=== Experiment 0 – samples_per_batch = 600 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 9604
Energy 0: -108.7451340647797
Batch 1 subspace dimension: 9604
Energy 1: -108.7451340647797
Batch 2 subspace dimension: 9604
Energy 2: -108.7451340647797
Batch 3 subspace dimension: 9604
Energy 3: -108.7451340647797
Batch 4 subspace dimension: 9604
Energy 4: -108.7451340647797
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 1292769
Energy 0: -111.94008269184971
Batch 1 subspace dimension: 1283689
Energy 1: -111.90964108773579
Batch 2 subspace dimension: 1285956
Energy 2: -111.61918103587362
Batch 3 subspace dimension: 1301881
Energy 3: -112.11324484954643
Batch 4 subspace dimension: 1301881
Energy 4: -111.60948309409991
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 1140624
Energy 0: -113.26509549780968
Batch 1 subspace dimension: 1129969
Energy 1: -113.25410141311288
Batch 2 subspace dimension: 1121481
Energy 2: -113.26183399427917
Batch 3 subspace dimension: 1096209
Energy 3: -113.31056596708696
Batch 4 subspace dimension: 1098304
Energy 4: -113.28203116572283
=== Experiment 1 – samples_per_batch = 800 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 9604
Energy 0: -108.7451340647797
Batch 1 subspace dimension: 9604
Energy 1: -108.7451340647797
Batch 2 subspace dimension: 9604
Energy 2: -108.7451340647797
Batch 3 subspace dimension: 9604
Energy 3: -108.7451340647797
Batch 4 subspace dimension: 9604
Energy 4: -108.7451340647797
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 2140369
Energy 0: -112.51727168237161
Batch 1 subspace dimension: 2244004
Energy 1: -113.26518925387896
Batch 2 subspace dimension: 2169729
Energy 2: -112.04629322434131
Batch 3 subspace dimension: 2178576
Energy 3: -112.14023905686373
Batch 4 subspace dimension: 2220100
Energy 4: -111.87776259849824
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 1982464
Energy 0: -113.3191668568732
Batch 1 subspace dimension: 2022084
Energy 1: -113.34106783990188
Batch 2 subspace dimension: 2036329
Energy 2: -113.31949820511863
Batch 3 subspace dimension: 1957201
Energy 3: -113.3227064069527
Batch 4 subspace dimension: 1979649
Energy 4: -113.33444862042249
=== Experiment 2 – samples_per_batch = 1000 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 9604
Energy 0: -108.7451340647797
Batch 1 subspace dimension: 9604
Energy 1: -108.7451340647797
Batch 2 subspace dimension: 9604
Energy 2: -108.7451340647797
Batch 3 subspace dimension: 9604
Energy 3: -108.7451340647797
Batch 4 subspace dimension: 9604
Energy 4: -108.7451340647797
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 3411409
Energy 0: -112.45510648564738
Batch 1 subspace dimension: 3301489
Energy 1: -111.89811835242475
Batch 2 subspace dimension: 3258025
Energy 2: -112.37038779001001
Batch 3 subspace dimension: 3392964
Energy 3: -111.86778609726969
Batch 4 subspace dimension: 3359889
Energy 4: -113.23606242669243
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 2958400
Energy 0: -113.37304053261217
Batch 1 subspace dimension: 2924100
Energy 1: -113.3381814894901
Batch 2 subspace dimension: 2975625
Energy 2: -113.36830669344843
Batch 3 subspace dimension: 2944656
Energy 3: -113.34623703652204
Batch 4 subspace dimension: 2944656
Energy 4: -113.34228018178595
=== Experiment 3 – samples_per_batch = 1200 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 9604
Energy 0: -108.7451340647797
Batch 1 subspace dimension: 9604
Energy 1: -108.7451340647797
Batch 2 subspace dimension: 9604
Energy 2: -108.7451340647797
Batch 3 subspace dimension: 9604
Energy 3: -108.7451340647797
Batch 4 subspace dimension: 9604
Energy 4: -108.7451340647797
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 4524129
Energy 0: -112.36944341019313
Batch 1 subspace dimension: 4713241
Energy 1: -113.24469042038862
Batch 2 subspace dimension: 4635409
Energy 2: -113.28712874549532
Batch 3 subspace dimension: 4631104
Energy 3: -112.37552643490679
Batch 4 subspace dimension: 4678569
Energy 4: -112.30090822945243
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 4157521
Energy 0: -113.36276499352192
Batch 1 subspace dimension: 4284900
Energy 1: -113.3785175436852
Batch 2 subspace dimension: 4251844
Energy 2: -113.36505824729713
Batch 3 subspace dimension: 4305625
Energy 3: -113.36782435540138
Batch 4 subspace dimension: 4260096
Energy 4: -113.36533292786831
=== Experiment 4 – samples_per_batch = 1400 ===
Starting configuration recovery iteration 0
Batch 0 subspace dimension: 9604
Energy 0: -108.7451340647797
Batch 1 subspace dimension: 9604
Energy 1: -108.7451340647797
Batch 2 subspace dimension: 9604
Energy 2: -108.7451340647797
Batch 3 subspace dimension: 9604
Energy 3: -108.7451340647797
Batch 4 subspace dimension: 9604
Energy 4: -108.7451340647797
Starting configuration recovery iteration 1
Batch 0 subspace dimension: 6330256
Energy 0: -112.48005975566662
Batch 1 subspace dimension: 6066369
Energy 1: -112.32119785649384
Batch 2 subspace dimension: 6255001
Energy 2: -112.83436524458222
Batch 3 subspace dimension: 6265009
Energy 3: -112.54947014782984
Batch 4 subspace dimension: 6155361
Energy 4: -112.45773309441128
Starting configuration recovery iteration 2
Batch 0 subspace dimension: 5631129
Energy 0: -113.3580204819709
Batch 1 subspace dimension: 5560164
Energy 1: -113.36852448354212
Batch 2 subspace dimension: 5607424
Energy 2: -113.36979135555231
Batch 3 subspace dimension: 5645376
Energy 3: -113.36601272012871
Batch 4 subspace dimension: 5664400
Energy 4: -113.37765089759762
To assess sample efficiency, you can compare the final SQD ground-state energy error as a function of the total number of measurement samples, with and without Fire Opal. For each point in the sampling schedule, the bar chart reports the absolute deviation from the CCSD reference energy (lower values indicate better performance) as a function of the maximum subspace dimension, following three rounds of classical configuration recovery. The subspace dimension indicates the classical computational resources allocated during the diagonalization step, larger dimensions require more classical computation but allow the solver to explore a richer basis of sampled configurations. Both execution methods are evaluated on the same backend with identical shot budgets to ensure a fair comparison.
The results reveal two principal trends. First, increasing the subspace dimension improves accuracy for both methods, since the classical solver gains access to a larger configuration space. Second, Fire Opal consistently achieves substantially lower error, approximately four times better across all tested subspace dimensions. This consistent improvement indicates that the use of Fire Opal enables the SQD solver to reconstruct a more accurate ground state from the same number of measurements.
plt.style.use(qv.get_qctrl_style())
samples = np.array(samples_schedule)
default_err = np.array(e_diff_default)
fo_err = np.array(e_diff_fo)
x = np.arange(len(samples))
width = 0.35
fig, ax = plt.subplots(figsize=(11, 8))
ax.bar(
x + width / 2,
fo_err,
width,
label="With Q-CTRL",
alpha=0.8,
edgecolor="black",
linewidth=1.5,
)
ax.bar(
x - width / 2,
default_err,
width,
label="Without Q-CTRL",
alpha=0.8,
edgecolor="black",
linewidth=1.5,
color="gray",
)
max_subspace_dim = 2 * (samples**2)
max_subspace_dim_millions = max_subspace_dim / 1e6
x_labels = [f"{val:.1f}" for val in max_subspace_dim_millions]
ax.set_xticks(x)
ax.set_xticklabels(x_labels)
ax.set_xlabel("Maximum sub-space dimension [million]", fontsize=16)
ax.set_ylabel("Ground-state energy error [Ha]", fontsize=16)
ax.set_yticks([1e-2, 5e-2, 1e-1, 1.5e-1])
ax.yaxis.set_minor_locator(mticker.MultipleLocator(0.01))
ax.grid(True, which="major", axis="y", linestyle="--", linewidth=0.8, alpha=0.7)
ax.grid(True, which="minor", axis="y", linestyle=":", linewidth=0.8, alpha=0.4)
ax.set_title(r"Formaldehyde (CH$_2$O) – Energy Error vs Samples", fontsize=20)
ax.legend(
title="Execution",
labels=["With Q-CTRL", "Without Q-CTRL"],
fontsize=16,
title_fontsize=16,
)
ax.annotate(
"",
xy=(0.62, 0.88),
xytext=(0.20, 0.88),
xycoords="axes fraction",
textcoords="axes fraction",
arrowprops=dict(arrowstyle="-|>", mutation_scale=24, linewidth=4),
)
ax.text(
0.4,
0.9,
"Increasing classical resources",
transform=ax.transAxes,
ha="center",
va="bottom",
fontsize=16,
)
ax.annotate(
"",
xy=(-0.14, 0.32),
xytext=(-0.14, 0.65),
xycoords="axes fraction",
textcoords="axes fraction",
arrowprops=dict(arrowstyle="->", mutation_scale=22, linewidth=4),
)
ax.text(
-0.16,
0.5,
"Lower is better",
transform=ax.transAxes,
ha="center",
va="center",
rotation=90,
fontsize=16,
)
with np.errstate(divide="ignore", invalid="ignore"):
factors = np.where(fo_err > 0, default_err / fo_err, np.nan)
best_idx = np.nanargmax(factors)
best_factor = factors[best_idx]
x_best = x[best_idx] + width / 2
y_best = fo_err[best_idx]
y_default = default_err[best_idx]
ax.annotate(
f"{best_factor:.1f}X",
xy=(x_best, y_best),
xytext=(x_best, y_default),
ha="center",
va="bottom",
fontsize=20,
arrowprops=dict(arrowstyle="->", mutation_scale=22, linewidth=4),
)
plt.tight_layout()
plt.show()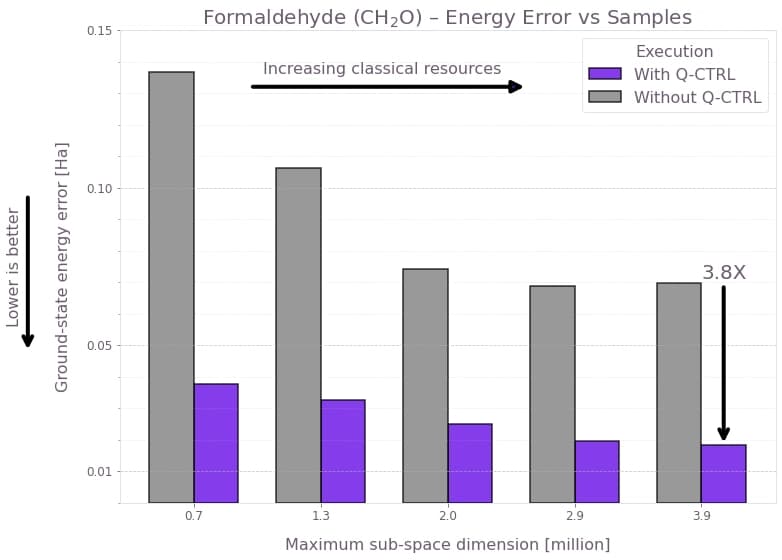
The following package versions were used to produce this notebook.
fo.print_package_versions()| Package | Version |
| --------------------- | ------- |
| Python | 3.11.14 |
| matplotlib | 3.10.8 |
| networkx | 3.5 |
| numpy | 2.3.4 |
| qiskit | 2.2.3 |
| qiskit-ibm-runtime | 0.43.1 |
| sympy | 1.14.0 |
| fire-opal | 9.1.0 |
| qctrl-visualizer | 9.0.0 |
| qctrl-workflow-client | 7.3.0 |
